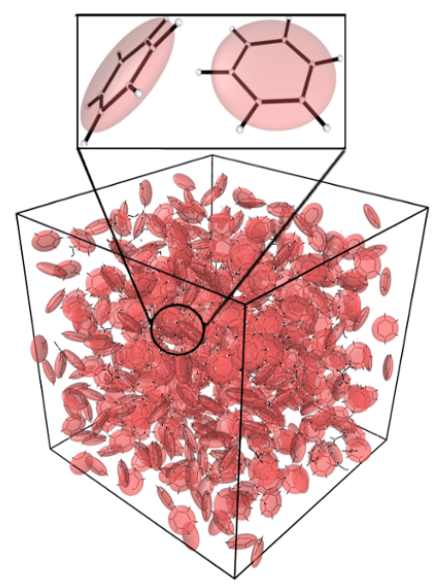
Coarse-Grained Molecular Dynamics
Many phenomena in soft condensed matter, such as supramolecular assembly of macromolecules, occur on length and/or time scales that are unfeasible to study theoretically or computationally if every atom in the system is represented explicitly. We are developing and applying new methods, using tools from statistical mechanics and machine learning, to simplify the representation of molecular systems in dynamical simulations by systematically eliminating unimportant degrees-of-freedom to improve computational efficiency without sacrificing accuracy. The end goal is to enable accurate simulations with molecular-level detail of large-scale dynamical processes that would be otherwise inaccessible by conventional techniques.
Organic Electronics
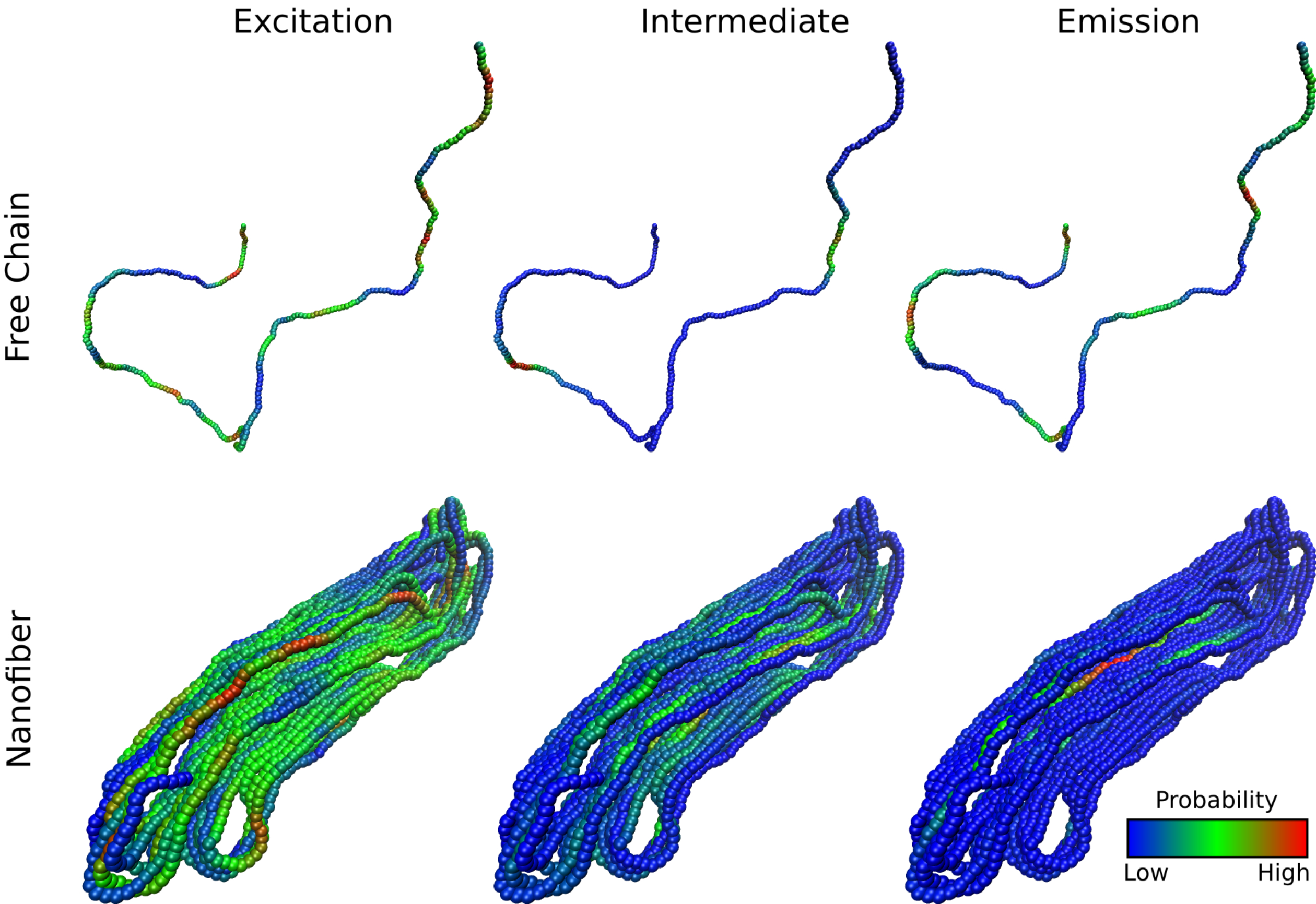
Carbon-based organic semiconductors are promising alternatives to conventional inorganic semiconductors such as silicon in electronic devices, especially when low cost, light weight, or flexibility is desired. They can also enhance inorganic semiconductor devices, e.g. by improving light harvesting in silicon solar cells. But organic-semiconductor devices are generally less efficient and their performance is often variable and hard to predict, largely because these materials are typically highly disordered, which can greatly impact device properties. We are using a combination of classical and quantum simulations to understand how structural and electronic properties of organic semiconductors can be controlled to enhance device performance.
Nanofluidics
The dynamics of liquids when they are confined by surfaces on the nano scale can deviate significantly from those at the macroscopic level. In particular, due to high surface-to-volume ratios, interface effects can dominate flow phenomena. We are using statistical mechanics and fluid dynamics to understand how molecular-scale surface interactions can be exploited to control nano-scale fluid flows, with a focus on applications to electrokinetic energy conversion processes.

